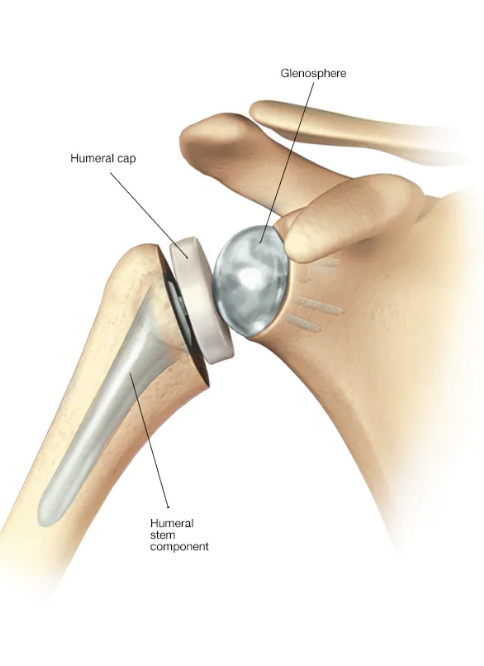
Unquestionably, the reverse total shoulder (rTSA) represents a major technological advance for the treatment of patients with complex shoulder disorders. The number of reverse total shoulders implanted in patients is growing rapidly due to (1) more surgeons using it, (2) its use in both older and young patients, and (3) its use to treat an expanding number of shoulder conditions.
Below are some comments on recent articles relevant to the rTSA.
FDA clearnance
The reverse total shoulder was first cleared for use in the United States by the Federal Drug Administration (FDA) twenty years ago. The FDA has the authority for approving use of orthopaedic implants by the 510(k) "clearance" mechanism, which bypasses the clinical trials required by the PreMarket Approval pathway which is used to assure the safety and efficacy of new medications (see How effective is the U.S. Food and Drug Administration (FDA) in assuring safety and effectiveness of shoulder arthroplasty implants? ).
Note below that the decision for 510(k) clearance of the reverse total shoulder was based on the device being "substantially equivalent" to a device previously cleared by the FDA.

While the anatomic total shoulder had been previously cleared by the FDA for use in the United States; there was no "substantially equivalent" reverse total shoulder that was "previously cleared" for use in the U.S. (most surgeons and patients would not conclude that the reverse total shoulder (below left) is "substantially equivalent" to the anatomic total shoulder (below right)).


A recent article authored by eight leading U.S. shoulder surgeons, Off-label use of reverse total shoulder arthroplasty: the American Academy of Orthopedic Surgeons Shoulder and Elbow Registry, observed that the FDA originally cleared the "on label" use of the rTSA for "grossly rotator cuff deficient joint with severe arthropathy or a previous failed joint replacement with a grossly rotator cuff deficient joint." They also pointed out that surgeons now use rTSA for "many off-label indications: osteoarthritis (OA) without rotator cuff tear (RCT), massive RCT without OA, proximal humerus fractures (PHFx), inflammatory arthritis (IA), and chronic glenohumeral joint dislocation (GHJD)".
They analyzed 3850 rTSA procedures reported to the AAOS registry from January 2015 to March 2021 "Only 24.4% of rTSA surgeries were performed for original on-label use (rotator cuff tear arthropathy). Off-label use of rTSA was seen in 75.6% of cases. When reviewing those rTSA done off-label, the majority (41.4%) were done for OA without RCT. Other off-label rTSA use included 15.1% for RCT without OA, 13% potentially off-label for PHFx, 4.6% for IA, and 1.6% for GHJD. Proportionally, off-label use is increasing over time while on-label use is decreasing."
These authors concluded that "Some implant manufacturers have expanded indications for rTSA without providing clinical data to support changing FDA approved indications for use. The incremental expansion of indications for use without supportive data - a practice known as predicate creep - is occurring with rTSA. Performing rTSA for off-label indications may create liability risk for surgeons and implant manufacturers. Device manufacturers should formally expand indications of use for rTSA with the FDA to be consistent with published literature and trends."
Indications
A common rationale for the use of the rTSA is pseudoparalysis - the inability of the shoulder to actively elevate the arm (see Pseudoparalysis in massive irreparable rotator cuff tears: what is it and why is it so important?). Shoulders without pseudoparalysis are often well treated with an anatomic shoulder arthroplasty, but those with pseudoparalysis are not (see What if the patient with an irreparable cuff tear and arthritis doesn't want a reverse total shoulder?).
With the expansion of indications for rTSA, it is apparent that a large percentage of patients receiving rTSA do not have pseudoparalysis. For example in Impact of accumulating risk factors on the acromial and scapular fracture rate after reverse total shoulder arthroplasty with a medialized glenoid-lateralized humerus onlay prosthesis, the preoperative active elevation for the 138 patients having postoperative scapular or acromial fractures complicating rTSA averaged 83.2 ± 41.8. A normal (bell-shaped) distribution of these values suggests that almost half of these patients may not have had preoperative pseudoparalysis.

Prosthesis design and position.
As suggested by these articles, the outcome of rTSA may be dependent on factors other than prosthesis geometry and implantation technique.
You can support cutting edge shoulder research that is leading to better care for patients with shoulder problems, click on this link.
Follow on twitter: https://twitter.com/shoulderarth
Follow on facebook: click on this link
Follow on facebook: https://www.facebook.com/frederick.matsen
Follow on LinkedIn: https://www.linkedin.com/in/rick-matsen-88b1a8133/
Here are some videos that are of shoulder interest
Shoulder arthritis - what you need to know (see this link).
How to x-ray the shoulder (see this link).
The ream and run procedure (see this link).
The total shoulder arthroplasty (see this link).
The cuff tear arthropathy arthroplasty (see this link).
The reverse total shoulder arthroplasty (see this link).
The smooth and move procedure for irreparable rotator cuff tears (see this link).
Shoulder rehabilitation exercises (see this link).